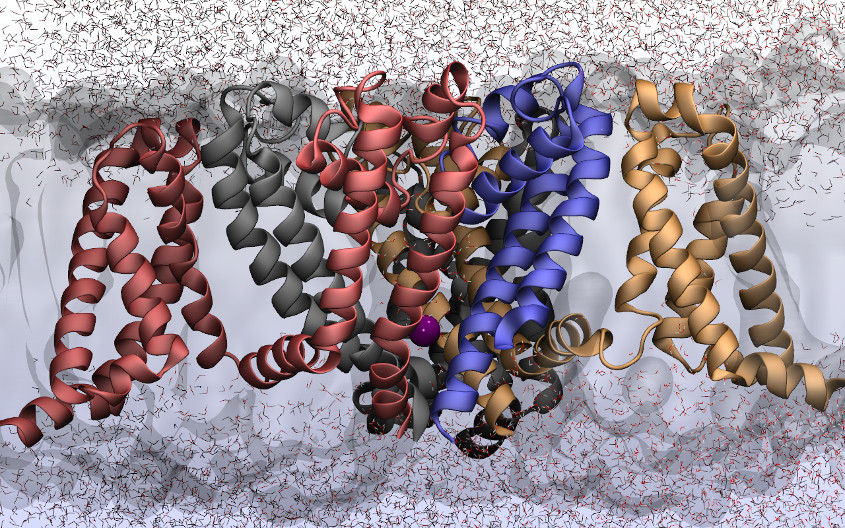
The NSF recently announced a new initiative to support the development of scientific software. One of my renderings (in this case, voltage gated sodium channel) was used as the press image! Unfortunately, the image is credited to Virginia Tech… Update: the attribution has been fixed. Thanks to all involved for the speedy correction.
Academic software is usually a horror to behold. As a big proponent of “you get what you pay for”, I’m excited to see what good this money can do for the molecular dynamics community.
A specific problem in the molecular dynamics community is, ironically, the abundance of software. Researchers can choose between Gromacs, Amber, Charmm, OpenMM, Hoomd, Acemd just to run their simulations. Forcefields and analysis are unfortunately usually tied to these “walled gardens”. I’m proud of the Pande lab’s attempt to unify some of these programs. OpenMM can read all sorts of different input file formats and MDTraj can read all sorts of different trajectory file formats. It’s easy to fall into the relevant xkcd trap when addressing these issues.
My workflow for starting a project
- Use charmm-gui for building a membrane. Charmm has the best system construction tools.
- Use Amber’s
tleapto set up force field parameters. Amber has nice forcefields. - Use OpenMM for running on Folding at Home GPUs. This is what FAH uses, and it’s easy to read amber
prmtopsinto OpenMM. - Use ParmEd to convert amber files to gromacs files.
- Use Gromacs to run on Folding at Home CPUs. This is what FAH uses.
- Use Gromacs to run on CPU clusters. It parallelizes really well.
- Use Amber to run on GPU clusters. It runs very fast.
Obviously, this would be easier if there was one package with all strengths, rather than many packages, each with their own strengths.